As síndromes medulares ocorrem quando partes da medula espinhal são danificadas, seja por traumas, doenças ou outras condições. Essa lesão interrompe as vias nervosas que transmitem informações entre o cérebro e o corpo, causando uma variedade de sintomas que dependem da área da medula afetada e da extensão do dano. As principais em provas são as neoplasias mieloproliferativas crônicas, relacionadas com mutações drivers no genes JAK2, CALR e MPL. Entre essas, podemos citar: policitemia Vera, Trombocitemia essencial, mielofibrose e leucemia mieloide crônica. Vamos explorar um pouco de cada uma delas?
Policitemia Vera
Por ser uma neoplasia mieloproliferativa crônica, observa-se uma proliferação clonal de células mieloides, afetando a hematopoiese. Por esse motivo, há aumento da massa eritroide (+ hemoglobina e hematócrito). A maioria dos diagnósticos ocorre de modo acidental, pela identificação da poliglobulia, em idosos e na população masculina.
Os pacientes podem apresentar sinais e sintomas como: cefaleia, alterações visuais, prurido “aqua gênico” (por exemplo após banhos quentes), eritromelalgia (queimação nas extremidades), tromboses, leucocitose, plaquetose e esplenomegalia.
No tratamento dessa síndrome, o objetivo é alcançar níveis de pelo menos 45% de hematócrito. Para isso, podem ser realizadas sangrias, hidroxiureia e associação com AAS.
Trombocitemia Essencial
Essa é uma síndrome que ocorre devido a ativação constante dos receptores de TPO – trombopoetina. Então, observa-se uma plaquetogênese intensa, contudo, não é comum quadros de anemia, poliglobulia e leucocitose. Essa é mais comum que a anterior e acomete, principalmente, mulheres e idosos. Um detalhe importante a se destacar a a apresentação de quase 90% de mutações no gene JAK2-V617F dos pacientes diagnosticados. Os sintomas dela incluem: plaquetose (com casos secundários de ferropenia, infecções, inflamação, esplenomegalia e trauma), esplenomegalia e sintomas vasomotores. O diagnóstico ocorre pela presença dos 4 maiores critério OU 3 critérios maiores e 1 menor:
| Critérios Maiores | Critérios Menores |
| Plaquetas > 450.000 /mm3 | ausência de causa secundária de trombose |
| Presença de JAK2, CARL e MPL | |
| Sem critérios para leucemia mieloide crônica, policitemia vera, síndrome mielodisplásica e mielofibrose | |
| Medula com aumento de megacariócitos atípicos |
O tratamento visa aliviar os sintomas e minimizar as complicações, visando alcançar níveis plaquetários entre 100 e 400 mil. Normalmente, são administrados interferon, hidroxiureia e AAS.
Mielofibrose
Entre as síndromes citadas, essa é a mais rara. Pode ser primária ou secundária a PV e TE. Ela decorre devido a mutações nos genes JAK2, CARL ou MPL e acomete principalmente pacientes com 67 anos ou mais. Podemos observar a presença de dacriócitos (hemácias em lágrima) no sangue periférico e uma possível evolução para leucemia mieloide aguda. Os pacientes são bastante sintomáticos, pois essa é uma doença de caráter extremamente inflamatório, e apresentam fadiga extrema, desconforto abdominal, tromboses,esplenomegalia maciça, anemia, leucocitose e plaquetopenia.
O diagnóstico ocorre pela presença de 3 critérios maiores e 1 menor:
| Critérios Maiores | Critérios Menores |
| Megacariócitos anômalos acompanhados de fibrose medular de grau maior ou igual a 2 | Anemia sem causa aparente |
| Presença de mutações JAK2 ou CARL ou MPL | Leucocitose maior que 11.000/mm3 |
| Ausência de BCR-ABL | Esplenomegalia palpável |
| Aumento de DHL | |
| Reação leucoeritroblástica |
O tratamento depende da condição do paciente. Se ele estiver em alto risco e for elegível, ocorre o transplante de medula óssea alogênico. Se estiver em risco intermediário, deve-se tratar de acordo com a sintomatologia (anemia, esplenomegalia, sintomas constitucionais). Em baixo risco depende: se assintomático apenas espera e assistência, se sintomático é preciso tratar conforme os sintomas.
Leucemia Mieloide Crônica
Essa síndrome ocorre devido à translocação que envolve os braços longos dos cromossomos 9 e 22, o que leva à fusão dos 2 genes e formação do BCR-ABL. Esse processo ativa constantemente a enzima TK, estimulando a desregulação da proliferação e maturação dos granulócitos. Essa síndrome representa de 15 a 20% das leucemias nos indivíduos adultos, sendo os diagnosticados, normalmente, homens maiores de 60 anos.
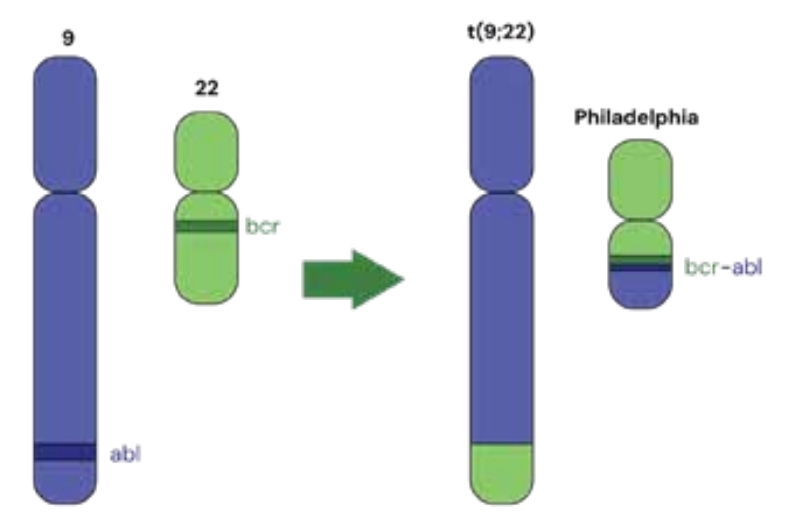
Os pacientes podem apresentar: leucocitose com desvio escalonado para esquerdo e basofilia (principal marco diagnóstico), esplenomegalia, fadiga e perda de peso. Porém, é importante pontuar que quase metade dos pacientes é assintomática até o diagnóstico. Aqueles sintomáticos podem passar por 3 fases: crônica (90% dos pacientes), acelerada (blastos de 10 a 19% na medula e evolução citogenética) e blástica (20% ou mais de blastos no sangue ou na medula, sarcoma mieloide e infiltrado de células blásticas extramedulares).
Para diagnosticarmos, precisamos nos atentar a diferenciá-la de reações leucemoides observando a presença dos sinais já expostos e do cromossomo Philadelphia. O tratamento envolve os inibidores TK!
Síndrome Mielodisplásica
Essa é uma neoplasia caracterizada pela hematopoiese clonal associada a citopenias. O risco de evolução para LMA depende das mutações envolvidas, motivo para acompanhamento constante do paciente! O risco aumenta se ele for exposto previamente a quimioterapia, radioterapia, benzeno e tabagismo. Ela é mais frequente em homens e a prevalência aumenta com a idade. O Diagnóstico se dá por observação de proliferação monoclonal de células tronco-hematopoiéticas, eritropoiese ineficaz, citopenias e anormalidades genéticas. Normalmente apresentam quadros que variam com a citopenia envolvida, mas podemos citar fadiga, infecções, sangramento e anemia (mais comum).
O tratamento dessa síndrome ocorre com o manejo dado a partida pontuação do risco:
- Alto: se elegível TCTH, se não são administrados hipometilantes.
- Intermediário: tratar de acordo com a sintomatologia, e se avaliar fazer TCTH.
- Baixo: se assintomático apenas esperamos, se sintomático são tratados os sintomas.
Anemia Aplástica
Representando uma doença de falência medular, essa síndrome tem caráter autoimune e causa pancitopenia em pacientes jovens. Ela é uma anemia macrocítica, com ausência de esplenomegalia, o que é um fator que já nos guia ao diagnóstico quando estamos investigando um paciente. Outros fatores que possibilitam o diagnóstico é biópsia de medula óssea (em busca de hipocelularidade global. Por fim, o tratamento dessa condição envolve a imunossupressão como conduta inicial, seguida por transplante de medula óssea como método curativo.
Seja um MedCoffer
Quer garantir a sua aprovação nas provas de residência médica? Então, conheça o Grupo MedCof! Aqui temos conteúdos de qualidade e especialistas que te ajudarão a alcançar a aprovação.